

快速將醫材進軍各國市場
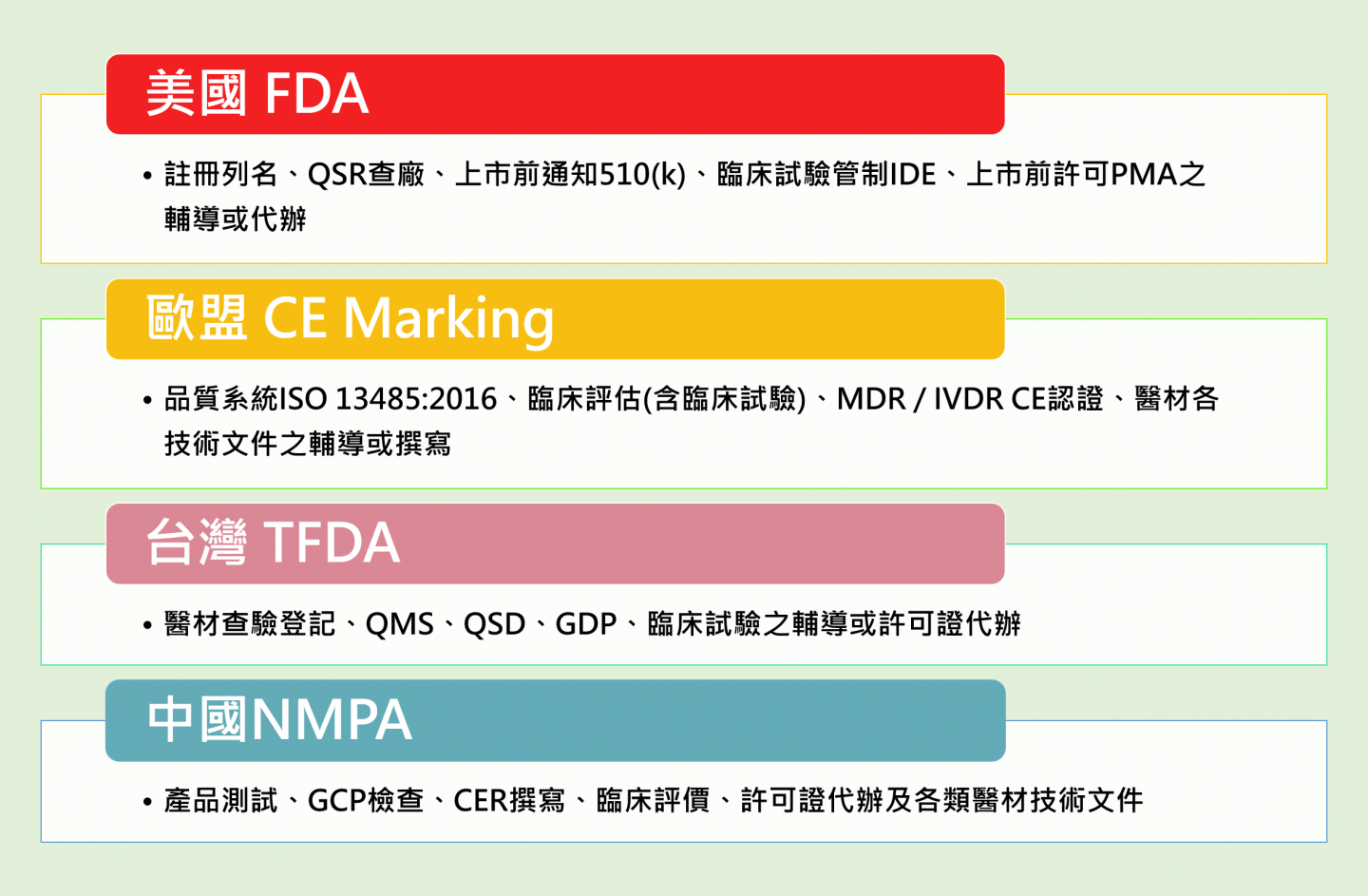
領証顧問提供專業的醫療器材法規輔導、撰寫技術文件、及許可證代辦服務,其範圍包含以下多國之醫材註冊輔導:- 台灣 TFDA : 台灣醫材許可證申請查驗登記(註冊)、代辦、醫材臨床試驗之申請輔導。
- 美國 FDA : 美國醫材510k申請註冊輔導(代辦),及PMA申請註冊輔導(代辦),及醫材臨床試驗申請輔導等。
- 歐盟 MDR CE : 歐盟MDR申請註冊輔導及體外診斷醫材IVDR申請註冊輔導 CE mark,及醫材臨床試驗申請輔導。
- 中國 NMPA(CFDA) : 中國醫材註冊申請輔導(代辦),及醫材臨床試驗申請輔導(CRO)。
- 日本 PMDA : 日本醫材註冊申請輔導(代辦),及 日本醫材品質管理系統查廠輔導。
- 東南亞(東協) : 新加坡、越南、馬來西亞等國醫材許可證(自由銷售證)申請註冊輔導(代辦),及醫材臨床試驗申請輔導。
- 澳洲 TGA : 澳洲醫材許可證澳洲註冊輔導(代辦),及醫材臨床試驗申請輔導。
醫材法規專業服務
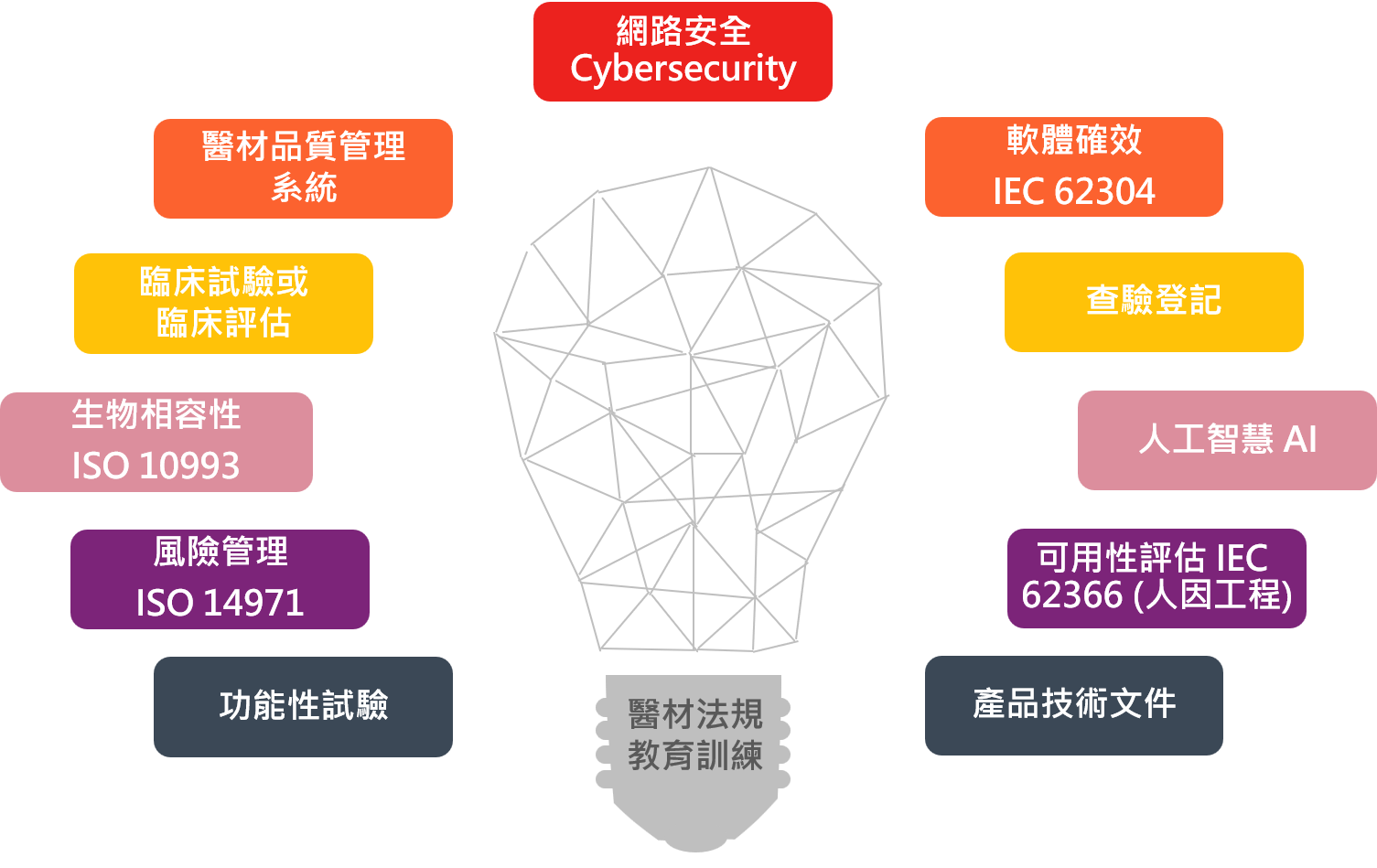
為了確保醫材的功效與安全性,提供以下醫療器材法規專業服務:
醫療器材註冊
醫材品質系統及環境系統輔導

醫材法規教育訓練
日本醫材註冊輔導
1. 日本醫材註冊分級

2. QMS稽核流程

Source: PMDA
3. 日本之PMD法案:
自2014年11月25日起,日本藥事法(PAL)經修訂,更名為「藥品、醫療器材、再生細胞治療產品、基因治療產品和化妝品品質保證、功效和安全的法案」(PMD法)。PMD法提供醫療器材、體外診斷試劑醫材、處方藥、藥品和化妝品、以及再生和細胞療法之產品在日本市場中的監管法律框架。PMD法的法律框架的管理和監督負責機構是日本衛生部,日本厚生勞動省(MHLW)和製藥和醫療器材局 (PMDA)共同管理。該機構執行藥品和醫療器材上市申請的科學評估,對其上市後的安全性進行監督。
4. 日本醫材法規要求 :
日本的醫療器材分類制度主要基於日本的醫療器材命名規範,該命名規範與美國或歐盟使用的分類系統有些區別。2005年4月起,第三方驗證機構(RCB)被允許評估二類醫療器材,並為在日本合法上市醫材出具銷售證明書。醫材在取得驗證之前必須證明符合日本產業標準(JIS),該標準定義了醫材之安全和性能要求。從2014年11月25日起,RCB還被允許評估第三級「類似」醫材,並提供與上市驗證服務。第三級「類似」醫材在獲得驗證之前,必須證明符合基本要求,這些要求與全球法規協調工作組(GHTF)指引調和。已在日本合法上市的實質等同之類似醫材也需要遵守該要求。
此外,如果製造場地執行以下活動,組織必須對其日本國外的製造場地進行註冊,並且獲得PMDA的製造廠註冊:
A : 設計和開發
B : 醫療器材的製造或滅菌
C : 體外診斷試劑的設計和開發或包裝
5. 日本醫療器材由厚生勞動省 (Ministry of Health, Labor and Welfare, MHLW)及藥品和醫療器材局(Pharmaceuticals and Medical Device Agency, PMDA)監督管理,厚生勞動省負責法規的管理及監督,藥品和醫療器材局負責對醫療器材進行技術檔審查制定權責及規定,以及評估和上市後監督及通報。再者,日本依照全球醫療器材法規調和會(GHTF)所定的風險等級分為四等級:
(1)第一等級:不須註冊(查驗登記),僅須向PMDA提交自我通知(self-notification)即可取得許可販售。
(2)第二等級及第三等級:多數第二等級醫材及極少數第三等級經MHLW授權的協力廠商註冊認證機構(RCB)審查,並取得PMDA批准。
(3)第四等級:需由PMDA審查並由MHLW批准。
6. 只有日本國內資格的當地機構才可進行進口及銷售,所以外國製造業者欲進入日本市場前,都需指定上市許可持有人(MAH)或指定上市許可持有人(DMAH)進行產品註冊,及與PMDA和RCB的聯繫窗口:
(1)上市許可持有人(Marketing Authorization Holder, MAH)
經由外國製造業者委託MAH為醫材註冊申請人,即為醫材註冊許可的所有者,負責進行產品註冊文件之提交、補件回覆、及申請證書轉讓,提交文件前無須取得外國製造業者的簽名。
(2)指定上市許可持有人(Designated Marketing Authorization Holder, DMAH)
DMAH是由外國製造業者委託於日本國內的代表,醫材註冊許可所有者為外國製造業者,DMAH負責進行產品註冊、補件回覆、及申請證書轉讓,提交文件前都須取得外國製造業者的簽名。
(3)日本醫材註冊之醫療器材相關標準
https://www.std.pmda.go.jp/stdDB/index_en.html
(4)日本醫材註冊之登記在案的驗證機構列
https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/kenkou_iryou/iyakuhin/tou roku/index.html
(5)日本醫材註冊之申請相關檔
https://www.pmda.go.jp/review-services/drugreviews/procedures/0004.html
東協醫材註冊輔導
1. 東協(東南亞)醫療器材指令 (ASEAN Medical Device Directive:2015)
(1)東協醫療器材風險分級
| 級別 | 風險等級 |
| A | 低風險 |
| B | 中低風險 |
| C | 中高風險 |
| D | 高風險 |
(2) CSDT的監管簡介
東協醫療器材指令 (AMDD) 附件4中建議使用東協共同提交檔案範本,該指令由東協地區十個國家簽署。CSDT旨在協調醫療器材上市前註冊所提交的文件,並簡化進入多個東盟國家的製造商的技術文件活動。此範本是國際醫療器材監管論壇 (IMDRF) 摘要技術文件(STED)的東協對應部分。
(3) 應用CSDT 範本進行上市前註冊CSDT範本適用於一般醫療器材及B級、C級、D類體外診斷醫療器材。
(4) CSDT 的語言要求AMDD要求CSDT技術文件以英文提供,除非東協成員國 (AMS) 要求以任何其他語言提供。
(5) CSDT技術文件的關鍵要素 -
A.執行摘要
B.用於證明符合性的基本原則和方法
C 設備描述
D.設計驗證與確認總結
E.臨床前和臨床數據(如有必要)
F.設備標籤
G.風險分析
H.製造商資訊應包括製造資訊、品質保證措施和滅菌方法的詳細訊息
(6) CSDT技術文件中應包含
A.有關醫療器材、器材的預期用途和使用適應症的介紹性描述訊息。
B.有關設備使用的資訊(如果有),例如目標患者群體、使用者概況(例如經過專門培訓的使用者)、特定疾病狀態或臨床狀況(例如對重症患者的持續監測)、作用模式(例如 對重症患者進行連續監測)。
C.如果醫療器材具有任何獨特或新穎的特徵或特性(例如奈米技術、包含動物或微生物細胞或組織),則必須提供描述。
D.產品所有者希望強調的與設備、其歷史或與其他批准設備(例如類似設備)或先前提交的關係相關的任何高級背景資訊或細節(提供提交的上下文)
E.設備的銷售歷史
F.醫療器材的註冊狀態(即已提交、未提交、待批准、批准、拒絕或回收)和批准的預期用途和適應症
G.每個參考機構的批准信副本。對於帶有CE標誌的設備,除了公告機構頒發的EC證書外,還必須提交產品所有者的歐盟符合性聲明
H.自醫療器材首次引入全球市場以來應報告的不良事件(AE)和現場安全矯正措施(FSCA)的摘要
I.對於「開放」的 FSCA,請提供產品所有者所採取的任何分析和/或糾正和預防措施的描述
J.如果迄今為止沒有發生不良事件或 FSCA,請提供產品負責人在公司信箋上提供的證明,證明自該設備在全球商業推出以來沒有發生不良事件或FSCA
K.基本原則符合性檢查表(EP檢查表)
L.符合性聲明 (DOC)
M.列出設備設計和製造(包括滅菌)過程中遵守的標準
掌握最新國際法規動態
醫材製造商最重要的是要知悉及遵循最新的法規動態,已經合法上市的醫材產品,亦須隨時注意新法規要求。
信任領証醫材法規顧問的法規專業與豐沛量能,可以幫助您解決各國醫材許可證註冊的複雜性,讓您可專心致力於醫材技術研發和市場開發,成為您取得各國許可証證的最佳後盾。
領証醫材顧問的目標
領証的目標是協助尊榮的客戶「100%領證」,至今仍未打破紀錄。我們團隊輔導產業從頭開始了解申請流程,視您需求客製化專屬貴司的臨床試驗與臨床前測試,並協助輔導或撰寫技術文件,以利有效益地進行醫材註冊及品質管理系統認證。

 加入領証生醫專頁
加入領証生醫專頁