

各國醫材要求UDI陸續上路
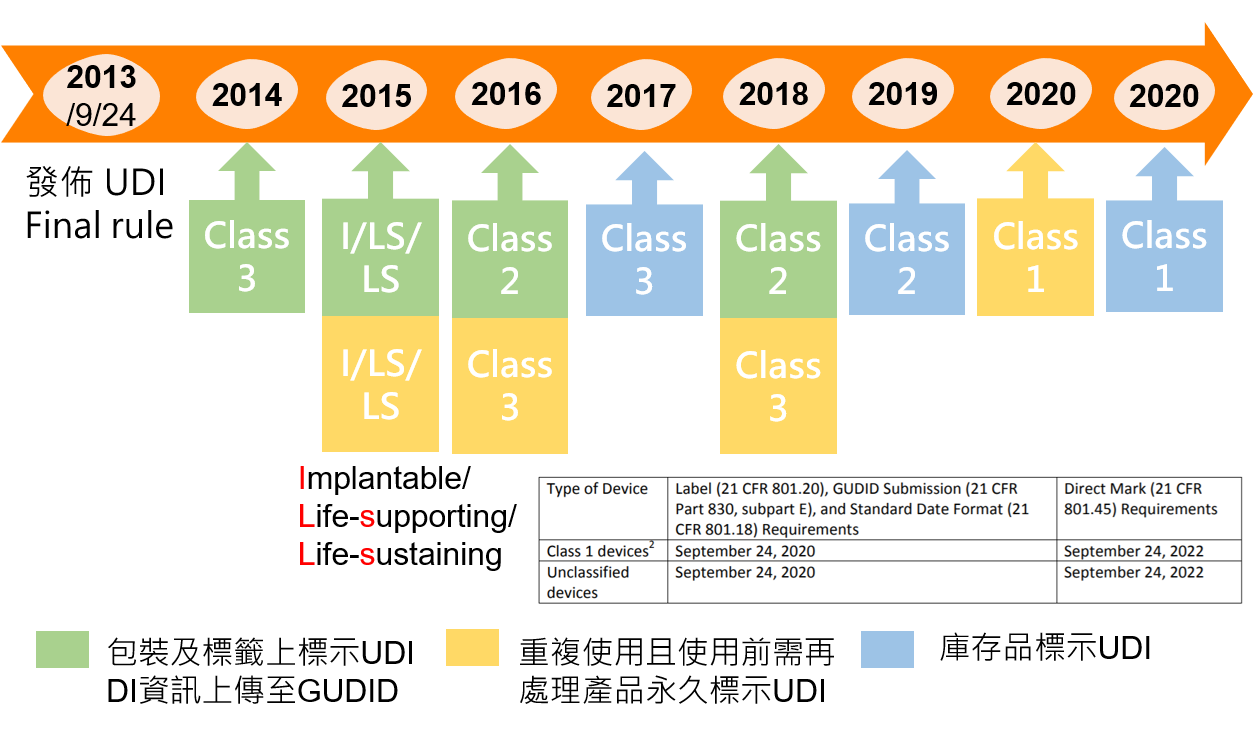
- 美國FDA公告自2020年起強制要求輸美產品皆須符合醫療器材單一識別系統(unique device identification (UDI) system)的要求。此外,FDA也同時要求輸美之境外廠商自2020年起,皆必須有自己的鄧白氏號碼。
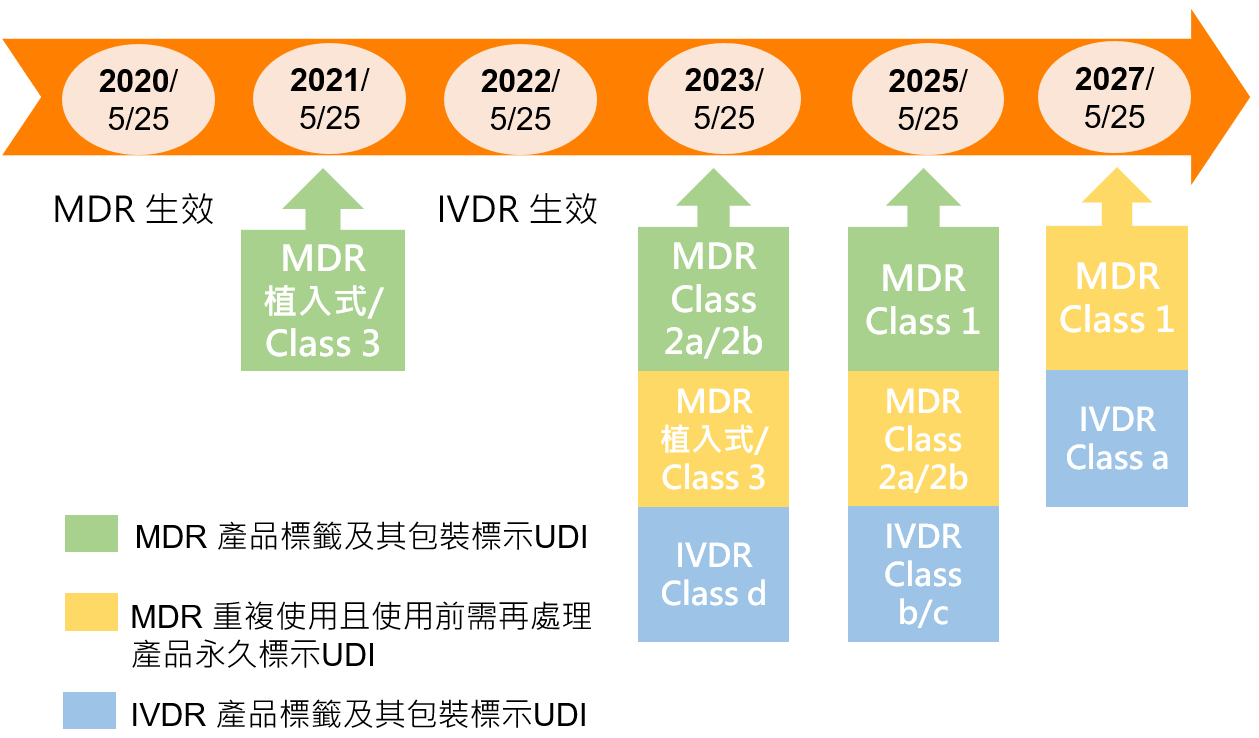
- 歐盟2020年亦將全面落實新的醫療器材法規, 強制要求醫材上市前,先在產品標籤上標示醫療器材單一識別條碼,才允許在歐盟市場流通。
- 台灣TFDA於2015年公告「醫療器材單一識別系統規範」,但只屬參考性質,至今醫材業者仍未將全數資訊上傳官方資料庫。醫療器材管理法於2019年1月15日由總統公布,依據醫療器材管理法第33條訂定”醫療器材標籤應刊載單一識別碼規定”草案,第二級及第三級醫療器材之單一包裝或器材本體上,應標示單一識別碼(UDI);而單一識別碼之產品對應資訊,應至醫療器材識別單一系統資訊管理平台登載(TUDID)。另外,第19條也明定醫療器材商及醫事機構應建立產品來源及流向之管理資料。政策計畫推動時程如下:
第一階段:2021年6月1日起製造之國產及輸入第3等級植入式醫療器材。
第二階段:2022年6月1日起製造之國產及輸入所有第3等級醫療器材。
第三階段:2023年6月1日起製造之國產及輸入第2等級醫療器材。
第二階段:2022年6月1日起製造之國產及輸入所有第3等級醫療器材。
第三階段:2023年6月1日起製造之國產及輸入第2等級醫療器材。
- 2019年8月27日,國家藥監局發佈《醫療器械唯一標識系統規則》,共18條,對醫療器械唯一標識系統的構成、唯一標識的技術要求、相關方責任等加以明確。規定國家藥監局負責建立醫療器械唯一標識系統制度,供公眾查詢,資料的真實性、準確性和完整性對於醫療器械的正確識別,強調了醫療器械註冊人/備案人應當對資料的真實性和準確性負責。已自2019年10月1日起正式實施。為落實醫療器械唯一標識系統內容之執行,於2020年12月底又公佈UDI數據資料庫之資料規格與管理信息系統之登錄入口。
參考資料:
TFDA https://www.fda.gov.tw/tc/newsContent.aspx?id=19214&chk=8f9b78b8-1c74-4c3c-972d- 899d566796db
FDA https://www.fda.gov/regulatory-information/search-fda-guidance-documents/unique-device-identification-policy-regarding-compliance-dates-class-i-and-unclassified-devices-and
NMPA https://www.nmpa.gov.cn/directory/web/nmpa/yaowen/ypjgyw/20190827103801436.html
EU https://ec.europa.eu/docsroom/documents/42641?locale=en

 加入領証生醫專頁
加入領証生醫專頁